如果有一天,医生跟你提起"要不要考虑参加一个临床试验"——你的第一反应是什么?
迟疑?害怕?还是有点不知所措?
不用觉得自己反应过度。临床试验这四个字,对大多数人来说都很陌生,陌生的东西让人不安,是再正常不过的事。
就比如:
这些情况,我见过很多次了。所以有了这个页面。
它不劝你参加,也不劝你拒绝。它只想把那些最常让人迷惑、害怕的事,用普通人能听懂的话说清楚:
简单说,药物临床试验是"在严格规则下、有人参与的研究"。它要回答几个问题:在人身上安全吗?这个药对相应的病到底有没有效?副作用是什么、发生率多高?
为什么必须做?因为任何一种药要在我国正式获批、被医生开给病人,都必须先在人身上经过严格的临床验证。我们今天能从医院药房或药店拿到的每一盒药,都是先有一群志愿者参与了临床试验之后,才走到我们手里的。
答:这是最常见、也最能理解的担心。但"小白鼠"这个比喻其实不准确——实验动物没有选择权、不知情、也没有人替它说话;而你作为受试者恰恰相反:你有权完整知道试验内容、自愿决定参不参加、随时可以退出,背后还有伦理委员会专门盯着保护你(见下一节"你的 8 项权利")。
而且,任何能进入人体试验的药,都已经做过大量实验室和动物研究、积累了相当的安全数据,才被允许用到人身上——不是把人"从零开始试"。所以你不是小白鼠,而是一位有知情权、有选择权、有人保护的参与者。
答:这是个很自然的想法,但要分两面看。
"新"确实意味着还没有最终定论——这正是它需要被研究的原因,我们不能、也不应该假装它一定比现有治疗好。
但"新"不等于"差",更不一定等于"未经验证"。临床试验里的"新药",其实涵盖多种情况:
- 全新化合物:确实是从未上市过的新成分;但在被允许进入人体试验之前,都经历了大量实验室研究和动物实验,有初步安全数据支撑——并非"直接拿人试"。
- 已在境外(如美国、欧洲)上市、但尚未在中国获批:在其他国家已有真实患者的使用经验和安全积累,并非真正意义上的"未知药物"。
- 改变剂型:原有成分已有充分数据,试验研究的是新的给药形式(如从口服片改为注射剂、或开发缓释版本),并非从零开始。
- 拓展适应症:这个药已批准用于某类疾病,现在研究它对另一类疾病是否同样有效——其安全数据相当充分。
- ……等等。
III 期试验通常会拿新药和现有标准治疗直接对比,正是为了客观回答"到底有没有更好"。所以更准确的说法是:新药是"有待验证的可能"。它到底好不好,要靠数据说话——而你有权在参加前,了解目前已有的数据到了什么程度。
答:这个担心是合理的,但要放在背景里看:
- 一个药能走到人体试验,前面已经做过大量动物和实验室研究,初步安全性是有数据支撑的。
- 试验从小范围、低剂量开始,一步步扩大(见扩展阅读:临床试验分期),正是为了尽早、在可控范围内发现问题。
- 参加期间,你会得到比平常门诊密切得多的监测;一旦出现不良反应,有明确的处理流程(见下一问"如果发生了不良反应,谁来管我?")。
- 万一真的因试验受到伤害,你也有保障:按规定,临床试验要为受试者投保;因试验造成的伤害,会获得相应的救治和补偿/赔偿(具体范围写在知情同意书里)。
答:不良反应有人管、有流程管,而且管得比平常更紧:
- 你被监测得比平时更密。 参加期间,你的身体状况会被定期、主动地检查和记录——很多变化甚至比常规看病更早被发现。
- 你会拿到紧急联系方式。 大多数试验会给你一个随时能联系到研究团队的电话,身体一有不对劲,你能第一时间找到人,而不是干等。
- 出现不良反应,有明确的处理流程。 研究医生有责任及时评估和处理;严重的情况(医学上叫"严重不良事件")还必须按规定上报给伦理委员会和药监部门。
- 与试验相关的损害,你能得到救治和补偿。 按 GCP 的要求,临床试验要为受试者投保;因试验造成的伤害,会获得相应的救治和补偿/赔偿(具体范围写在知情同意书里)。
- 必要时,会有人为你及时"叫停"。 这不只靠你的研究医生:如果医生判断继续参加对你个人不利,会让你停药或退出;在更高层面,申办方、药监部门、以及一些试验专门设立的独立数据监查委员会,只要评估发现继续下去对受试者不利,都有权叫停整个试验。你的安全,永远高于试验数据。
答:能,任何时候都能退出,不需要给任何理由。这是你最重要的权利之一(见"你的 8 项权利"第 3 条)。
- 退出不需要经过谁批准,也不会因此受到责怪或惩罚。
- 退出不会影响你今后在这家医院的正常就诊和应得的治疗。
- 医生可能会建议你退出前做一次简单的安全检查——这是为了你的健康,但做不做,仍由你决定。
你随时可以说"我不想继续了",这扇门一直开着。
答:通常不会,很多项目反而能减轻经济负担。
- 试验用药一般由申办方免费提供。
- 方案规定的相关检查、随访,通常也不另外收你的费。
- 不少试验还会给一定的交通、误工补偿。
答:保护你的隐私是法规的硬性要求,不是口头承诺。
- 你的身份信息会被编号、去标识化处理——在试验数据里,你通常是一个"编号",而不是"张三李四"。
- 能接触到你身份信息的人是严格受限的,且都负有保密义务。
- 试验结果对外发表或公示时(如 CDE 登记平台),呈现的是汇总数据,不会公开到某一个具体的人。
数据会被用于研究,但你的个人身份会被尽力隔离和保护。具体怎么保护,知情同意书里有专门一段,可以重点看。
不管你最终决定参加还是不参加,作为(潜在的)受试者,你都拥有几项写在法规里的权利。它们不是医院或药企"愿意给你"的,而是你本来就有的——任何一个合规的临床试验,都必须先保证这些权利。
依据:《药物临床试验质量管理规范》(GCP)、《赫尔辛基宣言》、《涉及人的生命科学和医学研究伦理审查办法》
如果你愿意进一步了解有没有适合自己的临床试验,可以从这三个正规渠道入手:
这是最现实、也最贴合你病情的第一步。医生最了解你的情况,能判断你是否可能适合某个试验,或者把你转介给正在做相关研究的科室。有想法,先开口问医生——比自己上网瞎找靠谱得多。
这是国家药监局药品审评中心的官方平台,所有在中国开展的药物临床试验都必须在这里登记,信息最全、最权威。
① 打开微信,在顶部搜索框搜索"临床试验查询";② 在结果里,认准带有「官网」标记的那一个,点进去。
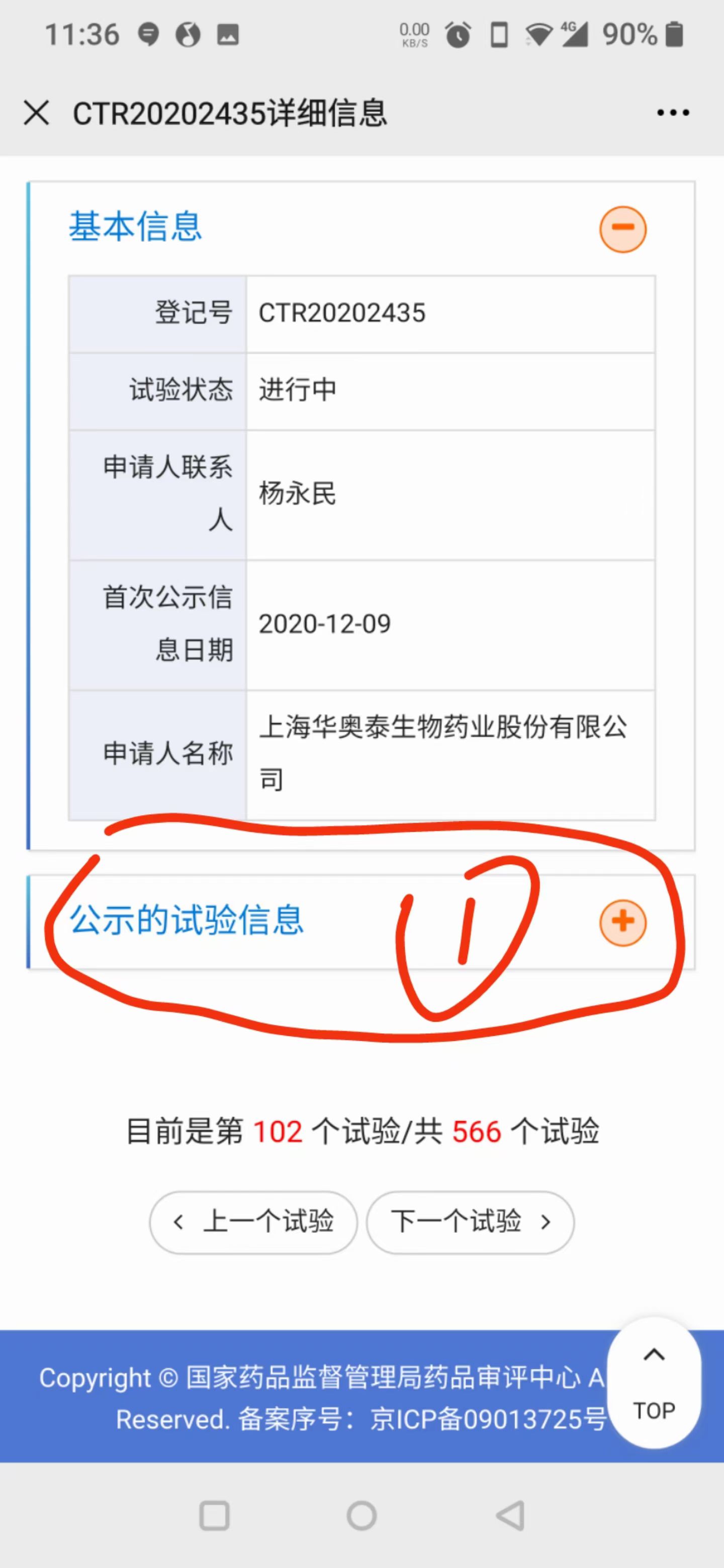
在搜索框里输入你的病名(即适应症,例如"肺癌"),点击"查询"。点击"试验状态"列进行排序,优先关注状态显示"进行中 招募中"的试验——这些是当前正在招募受试者的项目。

点击感兴趣的试验后,可以看到登记号、试验状态、申请人等基本信息。点击"公示的试验信息"右侧的"+"按钮,展开详细内容(适应症描述、入组条件、研究目的等)。
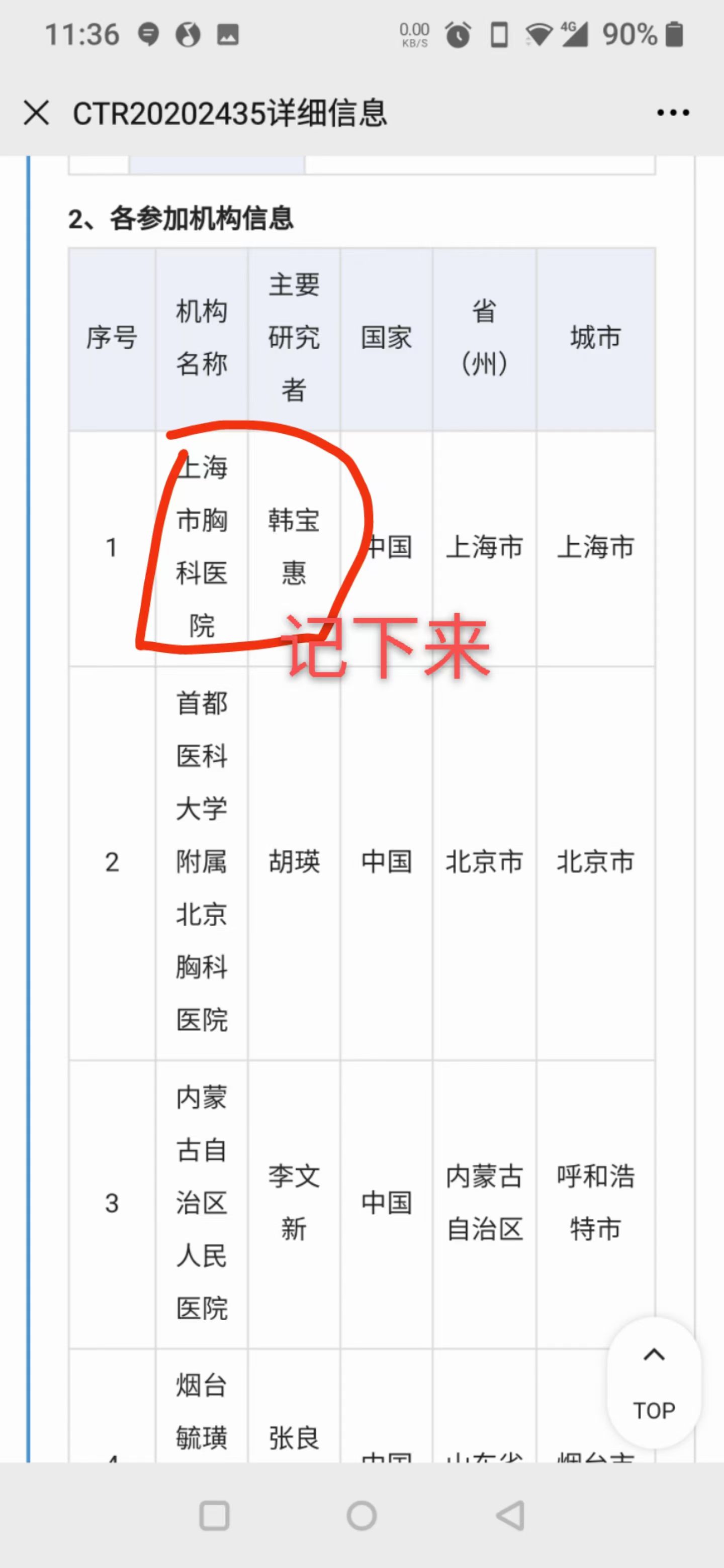
在"各参加机构信息"中,可以看到所有参加该试验的医院名称和主要研究者。找到离你最近的医院,把机构名称和研究者姓名记下来,直接去那家医院挂号或联系该研究者咨询。
正规试验的招募通知,常会张贴在医院相关病区或科室。住院时不妨多留意;看到感兴趣的,直接问你的管床医生或护士——他们能帮你确认这个试验是否正规、适不适合你。
万一你在参加过程中,遇到不公平、不舒服、被催促、或者对研究有疑虑的事,该找谁?
伦理委员会是一个独立于药企和研究医院的审查机构,依据 GCP 和《涉及人的生命科学和医学研究伦理审查办法》设立。它的职责:
- 在试验开始前,审查"这个试验值不值得做、是否充分保护了受试者"
- 在试验进行中持续监督,并接收受试者的疑虑和投诉
怎么联系它? 伦理委员会的名称和联系方式,会写在你签的《知情同意书》里(通常在专门一栏或最后一页)。签字前,找到它、记下来——这是你万一需要时的"后路"。
你不会"投诉无门"。这条独立的通道,一直为你留着。
临床试验通常分为四期,大致是这样一个由小到大、由摸索到验证的过程:
而这件事,不是任何一方能独立完成的——它需要五方共同参与,缺一不可:
缺一不可——任何一方都不可替代
五方之中,受试者贡献的是最不可替代的部分:真实的人体数据、真实的体感、真实的不良事件——这些,是临床试验最终能否回答"这个药有没有用、安不安全"的根本。
这一页里出现的一些词,这里用大白话解释,方便随时回查:
读到这里,谢谢你愿意花时间了解临床试验。
如果你正在考虑某个具体的试验,记住三句话:
这是你的身体、你的权利、你的选择。
免责声明
本页面为公益科普,旨在帮助公众了解药物临床试验的一般知识,不构成任何医疗建议,也不能替代医生的专业意见。
每一个临床试验的具体安排(用药、流程、费用、风险、补偿等)以该试验的方案和你签署的《知情同意书》为准;涉及你个人健康的任何决定,请务必咨询你的主治医生或专业人员。
参考法规与链接(均为官方来源)
- 《中华人民共和国药品管理法》(2019 修订)— 国家药品监督管理局
https://www.nmpa.gov.cn/xxgk/fgwj/flxzhfg/20190827083801685.html - 《药物临床试验质量管理规范》(GCP,2020 年第 57 号)— 国家药监局、国家卫健委
https://www.nmpa.gov.cn/xxgk/fgwj/xzhgfxwj/20200426162401243.html - 《涉及人的生命科学和医学研究伦理审查办法》(2023)— 国家卫健委等四部门
https://www.gov.cn/zhengce/zhengceku/2023-02/28/content_5743658.htm - 《药物临床试验登记与信息公示管理规范》(2020 年第 9 号)— 药品审评中心(CDE)
https://www.cde.org.cn/... - 《赫尔辛基宣言》(World Medical Association,现行 2024 年修订版)
https://www.wma.net/policies-post/wma-declaration-of-helsinki/ - 药物临床试验登记与信息公示平台(电脑端):https://www.chinadrugtrials.org.cn
- 同一平台(手机端):https://www.chinadrugtrials.org.cn/m_index.html